
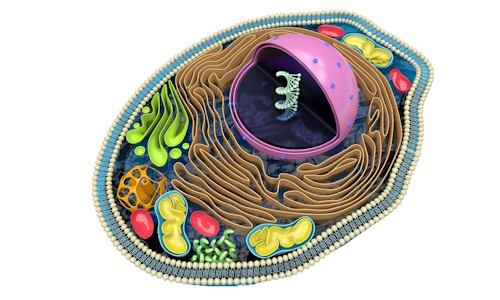
Qu'est-ce-que c'est ?
Sous l'appellation de « maladies lysosomales » sont regroupées une cinquantaine d'affections handicapantes de l'enfant et de l'adulte dont le point commun est une déficience génétique induisant un défaut de fonctionnement au niveau du lysosome. Situé au coeur de chacune de nos cellules, ce dernier a pour rôle de recycler des matières (appelées métabolites) issues du fonctionnement cellulaire.
Chaque maladie lysosomale implique un gène différent codant pour une protéine spécifique qui possède une fonction précise au sein du lysosome. Chaque protéine intervient au niveau d'un métabolite. Un défaut du gène entraîne, soit une non-production de la protéine correspondante, soit son dysfonctionnement. Le métabolite concerné n'est alors plus ou mal pris en charge dans le lysosome et s'accumule. Les cellules vont progressivement s'engorger, et les différents organes ne plus fonctionner normalement.
Liste des maladies lysosomales
Mucopolysaccharidoses
- MPS I (Hurler, Hurler/Scheie, Scheie)
- MPS II (Hunter)
- MPS III (Sanfilippo A,B,C et D)
- MPS IV (Morquio)
- MPS VI (Maroteaux-Lamy)
- MPS VII (Sly)
- MPS IX
Céroïdes lipofuscinoses
Pycnodysostose
Oligosaccharidoses et glycoprotéinoses
- Aspartyl glucosaminurie
- Fucosidose
- Alpha mannosidose
- Beta mannosidose
- Mucolipidose type II (I Cell)
- Mucolipidose type III (Pseudo Hürler)
- Mucolipidose type IV
- Sialidose
- Galactosialidose
Anomalies du transfert lysosomal
- Cystinose
- Maladie de Danon
- Maladie de Salla
Glycogénose type 2
Maladie de Pompe
Syndrome de Papillon-Lefèvre
Lipidoses
- Austin
- Fabry
- Farber
- Gangliosidose à GM1 (Landing)
- Gaucher
- Krabbe
- Leucodystrophie Métachromatique
- Niemann-Pick (A/B et C)
- Sandhoff
- Schindler
- Tay-Sachs
- Wolman
Symptômes
Peu à peu, des lésions apparaissent au niveau de différents organes (os, cœur, poumons, foie, rate, cerveau...) provoquant des troubles particulièrement graves et irréversibles. Le plus souvent, les signes révélateurs de la maladie sont absents à la naissance. Ils n'apparaissent qu'après une période d'évolution de quelques mois, de plusieurs années, voire même à l'âge adulte.
Origines
Dans une maladie lysosomale, pour une raison génétique, le lysosome n'assure pas sa fonction. Les métabolites s'accumulent progressivement dans les cellules et, par voie de conséquences, dans les tissus du corps de l'enfant ou de l'adulte malade et en perturbent leur fonctionnement.
Traitement et suivi
A ce jour, quelques médicaments, fruits de nombreuses années de recherche, sont disponibles pour des traitements par enzymothérapie substitutive (ou traitement enzymatique substitutif) ou par inhibition de substrat. Ils ont reçu une autorisation de mise sur le marché (AMM) pour les maladies suivantes :
- Gaucher
- Fabry
- Cystinose
- MPS I (Hurler, Hurler-Scheie, Scheie)
- MPS VI (Maroteaux-Lamy)
- Glycogénose type II (maladie de Pompe)
- MPS II (Hunter)
L'enzymothérapie substitutive : le but de ce traitement est d'apporter, par des perfusions intraveineuses régulières (toutes les semaines ou tous les 15 jours), l'enzyme spécifique (protéine) qui fait défaut dans l'une des maladies lysosomales. Cette enzyme est produite par génie génétique, purifiée et modifiée pour l'aider à atteindre les cellules de l'organisme qui ont le plus besoin de recycler le métabolite impliqué dans la maladie. Pour chaque maladie, il est nécessaire de développer la production d'une enzyme différente. Les cellules du cerveau sont très difficilement accessibles par ce type de traitement.
L'inhibition de substrat : le but de ce traitement est d'empêcher partiellement la production du ou des métabolites qui, par défaut génétique, ne sont pas recyclés par les cellules. Ce type de traitement permet de diminuer l'accumulation du métabolite et l'engorgement des cellules.
D'autres voies thérapeutiques, telle que la physiopathologie, sont également en cours de développement.
 0
0

 2
2